3-SUBSTITUIERTE
4-IMINO-TETRAHYDRO-CHINAZOLIN-2-ONE UND
4-IMINO-5,5-DIMETHYL-IMIDAZOLIDIN-2-ONE ALS IMINKOMPONENTEN FÜR CARBAMOYLIERUNGSREAKTIONEN
Sehr geehrter Herr
Vorsitzender, meine sehr geehrten Damen und Herren,
Bild 1 Allgemeine
Darstellung der Stoffklasse
seit einiger Zeit
beschäftigen wir uns mit "verkappten" Isocyanaten, die in
Modellspaltungsreaktionen wie die freien Verbindungen reagieren können[1].
Bild 2 Ketimin Imidate / Chinazolininone und Imidazolidinone
Im Rahmen dieser
Untersuchungen hatten sich die Umsetzungsprodukte zwischen Ketiminen oder
Imidaten und Isocyanaten im Kressewurzeltest nach Butula[2]als bisher
wirksamste Verbindungsklassen[3]
erwiesen. Da beiden Stoffklassen die C=N - Doppelbindung am abzuspaltenden
Molekülteil gemeinsam ist, suchen wir nach weiteren entsprechenden Strukturen
und wollten daher 4-Imino-tetrahydro-chinazolin-2-one und
4-Imino-imidazolidin-2-one als Iminkomponenten einsetzen. Zuerst möchte ich
über die Chinazolin-2-one sprechen.
An 3-Position substituierte
und an der Iminogruppe unsubstituierte Verbindungen wurden
erstmals 1960 von Breukink und Verkade[4] als aus
2-Aminobenzonitril und Isocyanaten mit Methanolatkatalyse erhalten. An der
3-Position substituierte und der Iminogruppe carbamoylierte Derivate
sind unseres Wissens bisher unbekannt.
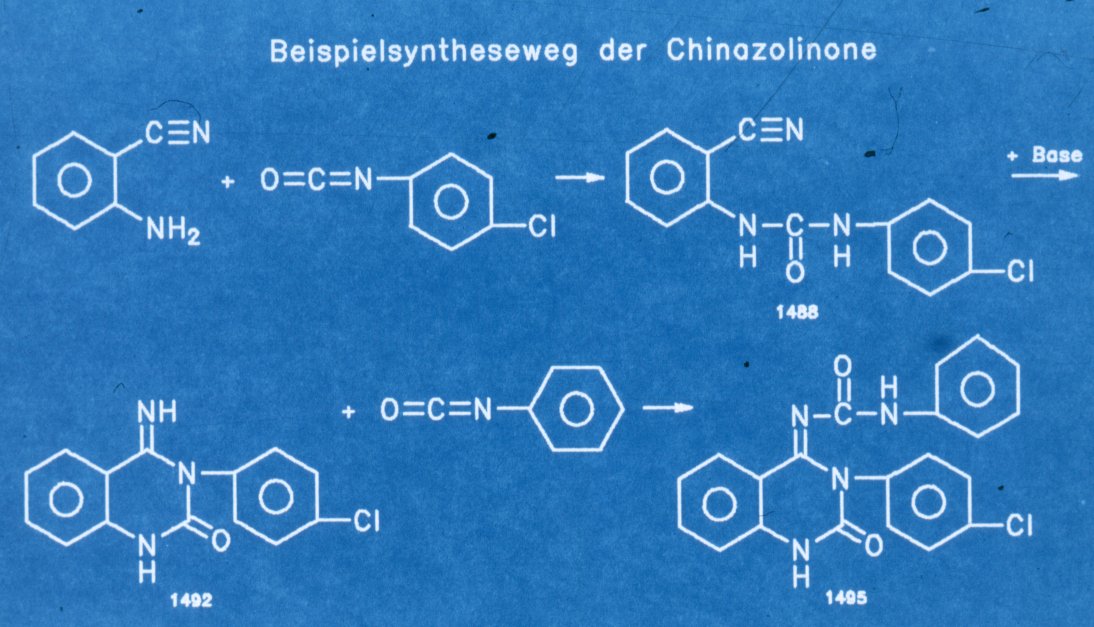
Bild 3
Darstellung von 1488 -->1492 --->1495
Wir führten die Darstellung
aus 2-Aminobenzonitril und entsprechenden Isocyanaten bei Raumtemperatur in
Ether, den Ringschluss unter Katalyse mit Piperidin oder Kaliumtertiärbutanolat
und die Umsetzung zum Zielprodukt erneut mit Isocyanaten durch. Ich möchte Sie
bitten , die mit 1492 gekennzeichnete Verbindung in Erinnerung zu halten, da
ich auf dieses im Folgenden näher eingehen möchte.
Bild 4
1492 in Deuteroaceton + Formel
Auf dem Dia sehen Sie einen
Ausschnitt des Aromatenbereichs des 1H Spektrums der Verbindung, der
mit einem vier-spin-System annähernd erster Ordnung und einem AA'BB' System die
angegebene Struktur zu bestätigen scheint. Da die Löslichkeit der Verbindung in
Aceton sehr schlecht war und wir auch 13Kohlenstoff-Spektren
aufnehmen wollten, wurden diese in DMSO aufgenommen. Und damit begannen die
Probleme.
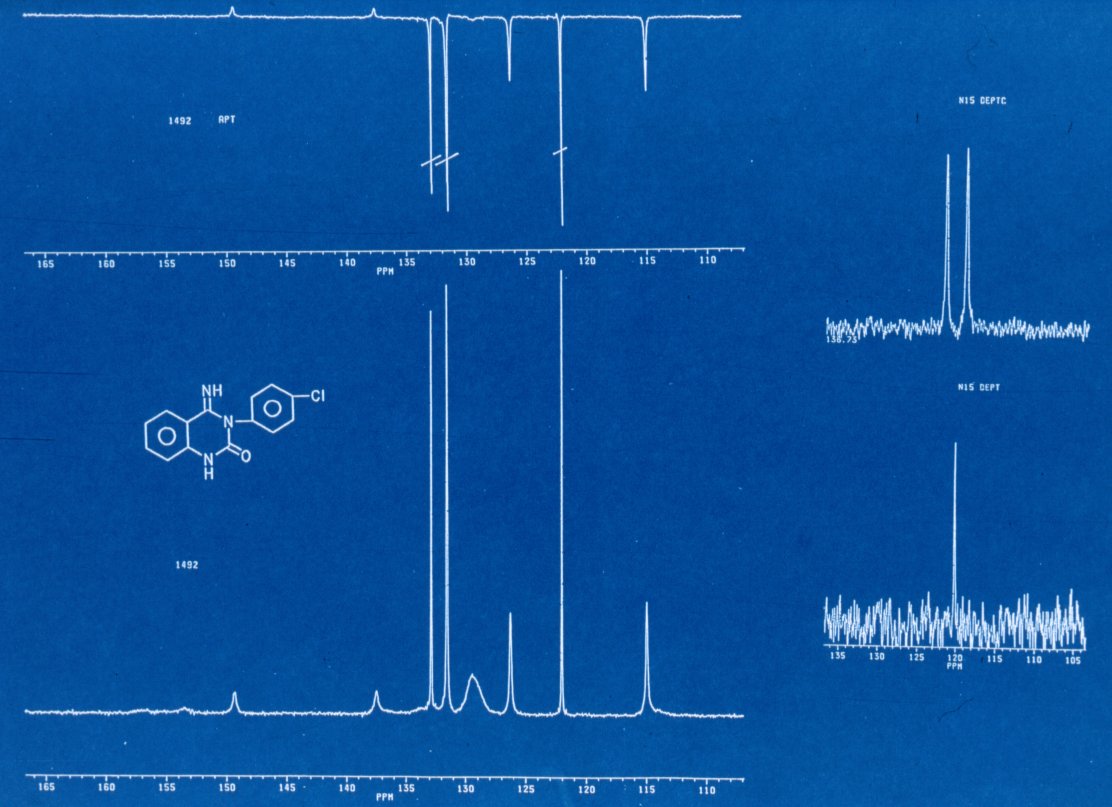
Bild 5 NMR
1492 (13C und 13C APT
15N DEPT) + Formel
Zu unserer Überraschung
zeigten sie nicht den kompletten Satz der erwarteten Signale [14] und diese
werden z.T. noch breit detekiert. [bestenfalls 8 - 10]
Beachten Sie dabei bitte
die Signale der quartären Kohlenstoffe im multiplizitätsselektierten Spektrum
[APT] . Es ist zu vermuten, daß sich vorwiegend diese an möglichen Tautomeren
beteiligen. Greift man dann in der Verzweifelung zum
15N-Spektrum
ist die Verwirrung komplett. In dem mit Hilfe der DEPT Technik aufgenommenen
Spektrum wären, methodebedingt, nach der Strukturformel je zwei Signale der
beiden Wasserstoff tragenden Stickstoffe zu erwarten. Tatsächlich beobachtet
man nur ein als [NH] Dublett auftretendes Signal.
Die erneut in DMSO
aufgenommenen 1H-NMR Spektren unserer Substanzen zeigten eine
zeitabhängige, reproduzierbare Veränderung der Spektren.
Bild
6 NMR 1492 (1H frisch, 5
Tage, H-H 2-D) + Formel
Im Bild sehen Sie erneut
den Ausschnitt der aromatischen Signale des 1H NMR Spektrums und
zwar einmal der frisch gelösten Substanz und einmal nach fünf Tagen.
Sie zeigen die erwarteten,
mit Sternchen gekennzeichneten, Signale für das vier-spin-System. Merkwürdig
sind hingegen die mit Punkten gekennzeichneten Signale des 4-Chlorphenylringes.
Anstelle eines einfachen AB-ähnlichen Systems sind verbreiterte Linien zu
beobachten, die auf vier separate Wasserstoffe hinweisen. Diese verändern sich,
in Abhängigkeit von der DMSO Qualität, [sprich dem Wassergehalt,] von einem
vier-spin System bis zu einem verbreiterten zwei-spin System und weisen
langsame Austauschreaktionen auf.
An dem gezeigten Ausschnitt
aus dem 2-D Spektrum kann man letzte Zweifel, ob es sich nicht doch um ein AB -
System handelt, beseitigen. Die Zuordnung der Kopplungssignale und die Größe
der chemischen Verschiebung von ca. 7 Hz ergibt, daß es sich um ein System mit
"gekreuzter" Kopplung handelt, wobei "gekreuzt" nur
geometrisch gemeint ist.
Welche
Erklärungsmöglichkeit sehen wir für unsere Beobachtungen ?
Bild 7 Dimroth - Umlagerung / Ergebnisse Taylor
In einer Arbeit von Taylor
und Mitarbeiter[5]
aus dem Jahre 1962 beschreiben die Autoren, daß sie aus
4-Imino-3-phenyl-tetrahydro-chinazolin-2-thion durch Dimroth -
Umlagerung auch die an der 3- Position unsubstituierten
N-substituierten 4-Amino-dihydro-chinazolin-2-thione erhalten hatten.
Die Dimroth Umlagerung wird üblicherweise als basisch katalysierte, thermisch
ausgelöste Ringöffnungs- und Recyclisierungsreaktion mit dazwischenliegender
Isomerisierung und "Platzwechsel" zwischen Imino- und Aminofunktion
beschrieben[6].
Eine analoge Umlagerung des 4-Imino-3-phenyl-tetrahydro-chinazolin-2-on
zu 4-Amino-dihydro-chinazolin-2-on gelang Taylor auch unter verschiedensten
basischen Bedingungen nicht, sondern führte nur zu Hydrolyseprodukten.
Wir hingegen glauben, dass die NMR Spektren unserer Substanz durch das im
folgenden Dia dargestellte Cyclotautomerengleichgewicht zu erklären sein
könnten.
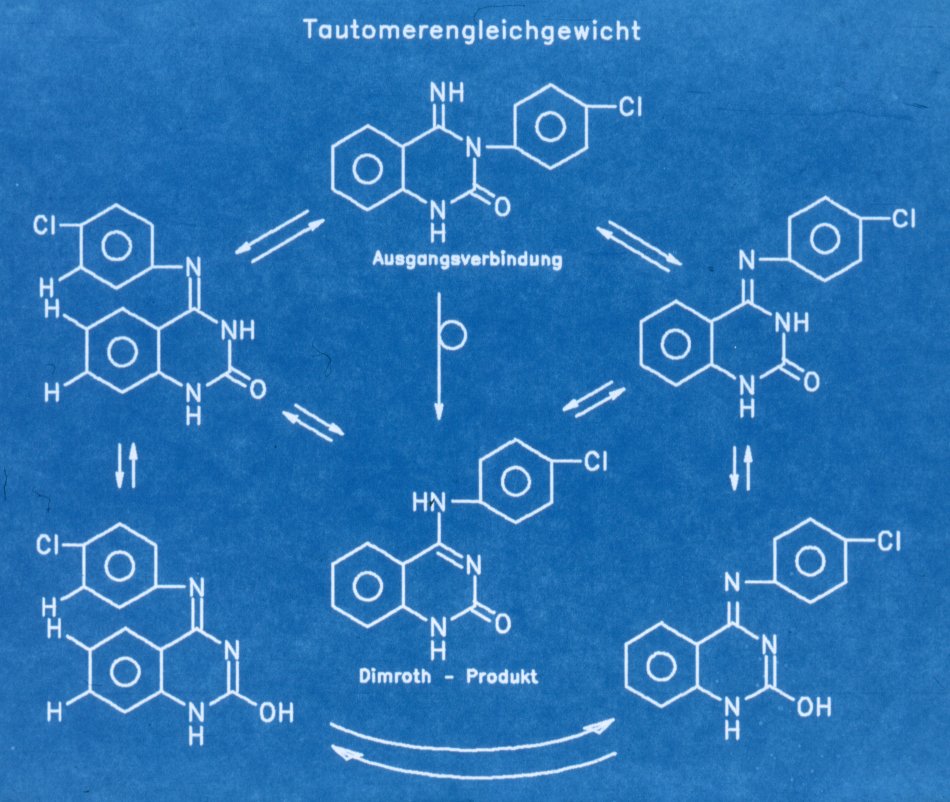
Bild 8 Tautomerengleichgewicht
Dieses
Tautomerengleichgewicht erklärt :
1) die beobachteten
mehreren breiten und labilen Wasserstoffsignale im 1H NMR,
2) die beobachteten vier
bzw. zwei Signale durch Einbeziehung des 4-Chlorphenylsystems in den Austausch
über E/Z Isomerie,
3) das Verhalten in den 13C
Spektren durch die wechselnde Umgebung
4) das eine scharfe
Stickstoffsignal in den 15N Spektren.
Wahrscheinlich handelt es
sich um den in allen Formen als NH gezeichneten Stickstoff, und allen anderen
gehen durch breite Koaleszenz im Grundrauschen unter. Das System erweist sich
also nicht als statisch und eindeutig, sondern das entsprechende
Dimrothprodukt ist zumindest im Gleichgewicht vorhanden. Die Veränderung der
1H
Signale zum zwei-spin System im nach Lagerung aufgenommenen Spektrum läßt eher
die Vermutung zu, daß das Dimrothprodukt den Endzustand darstellen könnte. Die
mituntersuchte Verbindung von Taylor und Mitarbeiter (1513) zeigt prinzipiell
gleiches Verhalten, wenn auch, durch das Fehlen der 4-Chlorsubstitution, nicht
so gut erkennbar. Da die Bedingungen im NMR Röhrchen doch stark von den
Bedingungen der Dimrothreaktion (Base, Temperatur) abweichen, ist es auch
möglich, daß eine Wanderung des Chlorphenylrestes vorliegt. Diese Ergebnisse
haben Konsequenzen für unser, gemäß den Angaben von Taylor, erwarteten
Wunschprodukts.
Bild 9
Versch. Struktur von 1495 aus 1492
Es könnten, bei Reaktion
als Dimroth - Produkt, auch die hier mit A und C bezeichneten Produkte
möglich sein, deren spektroskopische Unterscheidung sehr schwierig ist, da sie
nach Aussagen der NMR Fachleute im 1H
und 13C Spektrum zu ähnlich sind. Derzeit können wir Struktur B wohl
ausschließen. Im 15N Spektrum beobachten wir nur ein Signal, daß aber wie in der Vorstufe bei
120 ppm liegt. Andere Signale treten leider nicht auf. Wir sind dabei, das Problem weiter zu untersuchen.
Wie eingangs erwähnt wollten wir als zweite Stoffklasse Imidazolidinone
untersuchen.
Bild 10
Darstellung 1506 --->1508 --->1521
Nach gleicher Methode wie
bei den Chinazolinonen konnten wir auch Ringschlußreaktionen mit
N-Carbamoyl-"-amino-2-methyl-propionitrile zu
4-Imino-5,5-dimethyl-imidazolidin-2-onen durchführen. Auch bei dieser
Stoffklasse sind an der 3-Position substituierte und der Iminogruppe
carbamoylierte Derivate unseres Wissens bisher unbekannt, so daß wir die
dargestellte Reaktionsfolge durchgeführt haben. Bei diesen
"Fünfringanalogen" der Chinazolinone treten nach unseren bisherigen
Untersuchungen analoge Probleme wie im "Sechsringsystem" auf, auf die
ich hier jedoch nicht eingehen, sondern einen anderen Aspekt ansprechen möchte.
Nur das 2,2-dimethylierte Nitril cyclisiert, es gelang uns bisher nicht, die
Cyclisierungsreaktion auf analog gebaute Verbindungen anzuwenden.
Bild 11
Rechnerbild mit v.d. Waals-Radien
Da wir uns dieses Ergebnis
so nicht erklären konnten, haben wir versucht das Problem theoretisch anzugehen
und von den Strukturen an unserem Rechner, einem IBM 6150 mit dem Programm
MAD, Konformationsbetrachtungen
durchgeführt. Dargestellt sehen Sie die Raumerfüllung der Moleküle mit Hilfe
der van der Waals Radien. MAD verwendet zur Erstellung der Strukturen und zur
Energieberechnung Kraftfeldrechnungen. Dabei werden Terme für die Kompression,
Valenzwinkel, cross-term-streching, van der Waals - und Dipolwechselwirkungen
berücksichtigt. Erfahrungswerte wie gut das Programm Konjugationen berücksichtigt,
die bei vielen Programmen Schwierigkeiten machen, liegen uns noch nicht
ausreichend vor. Da die Struktur schwer zu erkennen ist,
Bild 12
Konformere, Farbdia vom Rechner
hier eine Darstellung ohne
die van der Waals Radien und zwar mit für den Ringschluß geeigneten
Atomabständen, in der jeweils energiegünstigsten Konformationen, sowohl der den
Ringschluß ergebenden (rechts) als auch nicht ringschließende (links)
Verbindungen. Oben drei Imidazolidinon-, unten, zum Vergleich zwei Chinazolinonvorstufen.
In allen Fällen ergeben sich dabei zum Bindungsschluß geeignete Konformere.
Betrachtet man aber die Rotationsenergiebariere um die entscheidende Bindung
zwischen "-Kohlenstoff und Harnstoffstickstoff zeigt sich das folgende
Bild.
Bild 13
Darstellung der Energiebarriere in Abh. vom Winkel
Dargestellt ist die
Energiebarriere in Abhängigkeit vom Torsionswinkel. Im Falle der
2,2-Dimethylsubstitution ist das Nitril aufgrund der sterischen Hinderung durch
die Dimethylstruktur in einer für die Reaktion günstigen Konformation (+/- 60
E) in einem "Energietal" praktisch fixiert. Das andere
"Energietälchen" von ca. 20 kcal/Mol, ist für den Ringschuß zum
Fünfring irrelevant, da hier die reagierenden Atome auf verschiedenen Seiten
stehen und maximale Atomabstände aufweisen. Bei der unsubstituierten bzw. der
monophenylsubstituierten Verbindung besteht dagegen keine solche
"energetische Fixierung". Die Energieunterschiede sind hier nur ca. 4
kcal/Mol. Die durch gegenseitige Behinderung der Aromaten entstehende hohe
Bariere am monophenyl System spielt keine Rolle, da der Aromat am
"-Kohlenstoff eine nicht behinderte Drehung in Gegenrichtung ausführen
kann.
Die beschriebenen
energetischen Verhältnisse könnten eine Erklärung für die beobachtete
Reaktivität sein.
Wir haben vor, der
Problematik durch weitere rechnergestützte experimentelle Untersuchungen
nachzugehen.
Damit bin ich am Ende
meines Vortrages.
Abschließend möchte ich dem
Leiter der NMR-Abteilung des Institutes für Anorganische und Angewandte Chemie
unserer Universität, Herrn Dr. Haupt, dafür danken, daß er unsere Verbindungen
so umfangreich untersucht hat[7]
und Ihnen, meine Damen und Herren, danke ich für Ihre Aufmerksamkeit.
Zur Diskussion :
1)
Tieftemperaturuntersuchungen ?
Dieses Verhalten lässt sich
auch durch Temperaturänderungen beeinflussen, doch leider findet man, aufgrund
der schlechten Lösungseigenschaften der Substanz, kein geeignetes Lösungsmittel
für Untersuchungen bei tieferen Temperaturen. Spektren auf Folie.
1a) Wiederspruch zu dem Ergebnis
Taylor. Was fand Taylor ? Versch.
Hydrolysen in sauren und basischen.
Damals selbstverständlich
nicht die gleichen technischen Möglichkeiten wie heute bestanden.
2) Wie klärt man Struktur und
Mechanismus ? Weitere Untersuchungen, wie z.B. eine der am Iminostickstoff 15N
gelabelten Substanz, zu denen wir leider bisher nicht über entsprechendes
Material verfügen, müssten hier zur Absicherung folgen.
3) Struktur des
Wunschproduktes ? Untersuchungen der Struktur 1495 über Massenspektroskopie
anzugehen, wurden begonnen. Spektrum auf Folie. Hinweis auf
Dimroth-Produkt.
4) Struktur der
Imidazolinone ? Wahrscheinlich auch Dimroth. Trennung in Arbeit. Spektren
auf Folie.
5) Warum nicht andere
Berechnungen ? Eine MNDO-Rechnung würde bei unserem Problem nicht weiterhelfen,
da die Ergebnisse für Nitrile nach neuerer Literatur zu stark abweichen[8].
Zu anderen (aufwendigeren) Verfahren [ab initio) fehlen uns die Möglichkeiten.
6) Wie wirken die
Substanzen ? Weil uns die die chemisch theoretischen Probleme der Strukturen
doch mehr als erwartet beschäftigten, kann ich Ihnen hier leider noch nichts
über die Wirkung der carbamoylierten Verbindungen berichten da wir sie, solange
ihre Struktur nicht geklärt ist, noch nicht untersucht haben. Hätten wir uns
auf die Literatur oder unser erstes
1H Spektrum in Aceton verlassen,
hätte ich Ihnen wohlmöglich Wirkungen beschrieben, die einer völlig anderen
Struktur zuzuschreiben sind.
[1]. H.G. Schweim und S. Jürgens, Arch. Pharm.
(Weinheim) 320, 844 (1987).
[2]. L. Butula, Pharmazie, 33, 430 (1978).
[3]. H.G. Schweim, Die Pharmazie, 44, 319
(1989).
[4]. K.W. Breukink und P.E. Verkade, Recueil Trav.
chim. Pays-Bas 79,
443 (1960).
[5] E.C. Taylor u. R.V.
Ravindranathan, J.org.Chem. 27, 2622 (1962)
[6] D.J. Brown in Mech.
of Mol. Migrations (Ed. B.S. Thyagarajan, Vol 1 (1968)
[7]. Alle Spektren wurden mit einem
BRUKKER AM 360 aufgenommen.
[8]. J.P. Steward,
J.Comp.Aid.Mol.Design. 1, (1990)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}