Beziehungen
zwischen chemischer Struktur und Physiologischer Wirkungen von Arzneistoffen
(Kurz : Struktur –
Wirkungsbeziehungen)
Meine sehr geehrten Damen
und Herren,
das Thema möchte ich Ihnen am Beispiel die Geschichte
und Entwicklung von Chemotherapeutica an Beispielen aus der Arzneimittelchemie
bis in die Gegenwart darstellen.
Bevor man sich über die Struktur-Wirkungsbeziehungen
unterhält, muss man zuerst einige Begriffe klären.
Unter "Infektionskrankheit" versteht man
heute alle Erkrankungen, die durch Viren, Protozoen, Bakterien, Pilze, Würmer
oder durch innere und äußere Parasiten hervorgerufen werden können.
Während man früher Arzneistoffe zur Bekämpfung von Infektionskrankheiten
je nach ihrer Gewinnungsart in "Antibiotica" und
"Chemotherapeutica" unterteilte, faßt man heute unter diesem Begriff
alle chemisch definierten, niedermolekularen Wirkstoffe zusammen, die zur
Prophylaxe und Therapie von Infektionskrankheiten und malignen Tumoren
eingesetzt werden. Gleichgültig, ob es sich dabei um Naturstoffe, abgewandelte
Naturstoffe oder Synthetica handelt.
Warum gerade Chemotherapeutica als Beispiel ?
Erstens weil diese Arzneistoffklasse mich von Beginn meiner Beschäftigung mit pharmazeutisch-chemischen Fragestellungen bis heute am meisten fasziniert.
Zweitens weil ich, wie viele andere auch, im Gegensatz
zu einer Reihe von Stoffen, deren therapeutischer Wert zweifelhaft ist,
Chemotherapeutica zu den unverzichtbaren Arzneistoffen rechne.
Drittens, weil es auf diesem Gebiet für die
Pharmazeutische Chemie zwar große Erfolge gab, aber immer noch große
Herausforderungen bestehen.
Legt man die Definition der
Weltgesundheitsorganisation zu Grunde und betrachtet nur Einzelsubstanzen, sind
ca. 1/3 aller Arzneistoffe Chemotherapeutica. Eine Behandlung aller Stoffe ist daher hier
unmöglich. Ich kann und werde ihnen also nur eine subjektive
Auswahl von Beispielen geben.
Die Wirkung beruht dann auf spezifischen, teils reversiblen,
teils irreversiblen Wechselwirkungen, wie z.B. chemische Umsetzung zu
kovalenten Bindungen, hydrophoben Wechselwirkungen oder Ionenbeziehungen zu dem
Rezeptor.
Dieser Wirkmechanismus gilt besonders für
Chemotherapeutica, bei denen es sich ja im Idealfall um Stoffe mit selektiver
Toxizität im Sinne der Ehrlichschen "Zauberkugel" für den
"Fremd"-Organismus handelt.
Dia. B1 [ß-Lactame/Ala-Ala Vergleich]
Gestützt wird diese Annahme durch die
Konformationsähnlichkeiten zwischen dem den ß-Lactamen zugrunde liegenden
Dipeptid aus D-Cystein und D-Valin und dem eigentlichen Substrat der
Carboxypeptidase, einem Peptid mit der Teilstruktur des D-Alanyl-D-alanin als
reaktivem Zentrum. Dies ist in der
Computersimulation der Partialstrukturen gut darstellbar.
Dia.
B2
[Computersimulation ß-Lact./Ala-Ala]
Der Rechner stellt Kohlenstoff blau, Wasserstoff weiß,
Sauerstoff rot, Schwefel gelb und Stickstoff violett dar.
Mit dieser kurzen Erwähnung möchte ich jedoch die
ß-Lactame und ihre Weiterentwicklungen aus meinem Vortrag ausklammern, da sie
doch eine recht komplexe und schwierige Chemie besitzen. An den Anfang der Besprechung von Beispielen möchte
ich eine alte, aber dennoch hoch aktuelle Substanz stellen, die einen Teil der
Schwierigkeiten der Arzneimittelentwicklungen deutlich macht.
Dia. B3 [Wirkm. des Cisplatin]
Bei dem Krebschemotherapeuticum Cisplatini
handelt es sich in vieler Hinsicht um einen "Exoten" im Arzneischatz.
So sind z.B. Anorganika nur zu ca. 10 % unter den Arzneistoffen vertreten und
ihre anwendungsbezogene Bedeutung ist noch geringer. Doch die
"Anorganica" sind z.Zt. "im Kommen". In neuester Zeit
werden zum Beispiel umfangreich Goldverbindungen
[Auranofin] auf ihre Eignung zur Therapie der "Volksseuche"
Rheuma untersucht. Der Wirkmechanismus des Cisplatini ist weitgehend
geklärt. Wirkstoff ist, wie im INN-Namen[3] angedeutet,
die cis-Form. Die trans-Form ist praktisch unwirksam. In Abhängigkeit von der
Chloridionenkonzentration im Gewebe bzw. der Körperflüssigkeit wird aus
Cisplatini der 2+ geladene diaqua-diammin-[1]
Komplex, der als Wirkform anzusprechen ist. Mit Isotopenmarkierungen [192Pt,
195Pt] konnte ein Wirkmechanismus nach dem "crossing link"
Prinzip unter kovalenter Fixierung der DNA über Platin wahrscheinlich gemacht
werden[2].
Damit wird eine Reduplikation der Erbsubstanz und damit eine weitere Vermehrung
der entarteten Zellen verhindert. Im Idealfalle wird so das weitere Krebswachstum
gestoppt. Auf Einzelheiten und Probleme der Pharmakologie des
Stoffes, seine Nebenwirkungen auf gesunde Zellen [Niere] sowie
Resistenzentwicklungen möchte ich hier nicht eingehen. Um, wie beschrieben wirken zu können, muß sich die
Wirkform zwischen die DNA-Stränge quasi 'drängen' können.
Der Abstand zwischen den Basenpaaren
Cytosin und Guanin bzw. Adenin und Thymin beträgt ca.
280 - 300 pm.
"Dazwischen zu
passen" ist für ein Molekül mit Platin als Zentralatom [Atomradius ca.138
pm] auf den ersten Blick nur schwer vorstellbar.
Dia. B4 [Computersimulation
Cisplatin und Diaquakomplex]
Der Nachbau der Moleküle am Computer (Platin blaugrün,
Chlor gelbgrün Stickstoff blau, Sauerstoff rot und Wasserstoff weiß), zeigt
jedoch eindrucksvoll die Änderung der Molekülgröße zwischen Transportform,
Moleküldurchmesser ca. 600 pm, [Atomradius Pt 1.38 Å, Cl 0.99 Å, N 0.7
Å, H 0.3 Å] und Wirkform, Moleküldurchmesser ca. 300 pm,
[Ionenradius Pt2+ 0.52 Å, O 0.66 Å] und macht so den geschilderten
Sachverhalt viel anschaulicher. Die
Substanz selbst ist schon 1845 von Peyrone[3]. M. Peyrone, Ann. 51, 1 (1845) synthetisiert, und
bemerkenswerterweise auch schon intuitiv mit richtiger Struktur beschrieben
worden.
Lange Zeit war die Verbindung, wegen des Vorliegens in
der cis-Form, nur von chemisch-theoretischem Interesse. Ihr Nutzen für die
Krebschemotherapie wurde erst 1965 durch Rosenberg[4][4],
also 120 Jahre nach der Erstsynthese, erkannt. Seit dieser Entdeckung hat eine stürmische Entwicklung
auf dem Sektor der "Organoplatinverbindungen" mit dem Anwendungszweck
"Chemotherapie maligner Erkrankungen" eingesetzt. Die therapeutischen Erfolge dieser Substanzen, z.B.
beim Hodentumor sind beeindruckend, bei anderen, weit mehr Opfer fordernden
Krebsformen wie z.B. Lungenkrebs, sind sie bisher leider enttäuschend.
Von einem "Sieg" über den Krebs, auch mit
weiterentwickelten Organoplatinverbindungen,
sind wir nach wie vor weit entfernt.
Das am Beispiel Cisplatini augenfällige
Dilema der Arzneimittelchemie liegt darin, unter Umständen hoch wirksame
Substanzen schon seit Jahren in der Hand zu halten ohne es zu wissen,
weil nicht jede neu synthetisierte Substanz auf jede denkbare Wirkung getestet
werden kann. Soweit dieses Beispiel für einen im wesentlichen
Zufallstreffer. Heute unternimmt man, auch aus ökonomischen Gründen, eine
Arzneistoffneuentwicklung kostet nach Angaben der Pharmazeutischen Industrie
bis zum Handelspräparat ca. 100 Mio DM,
Anstrengungen, um weg vom Zufallsbefund zu rationelleren Methoden der
Arzneistofffindung zu gelangen.
Welche Wege zu neuen Stoffen wurden historisch oder
werden noch heute erprobt ?
Dia.
B5
[Tabelle mit pot. Zugangswegen zu Arzneistoffen]
Einer der ältesten Wege zu Arzneistoffen ist die
Nutzung von Naturstoffen. Der Weg zu ihnen ist sicher über Selbst- und
Fremdversuch direkt am Menschen, oder über die Beobachtung von Tieren gefunden
worden. Das wird sicher nicht ohne dramatische Irrtümer abgegangen sein. Man
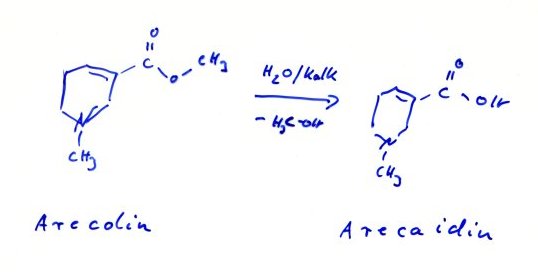
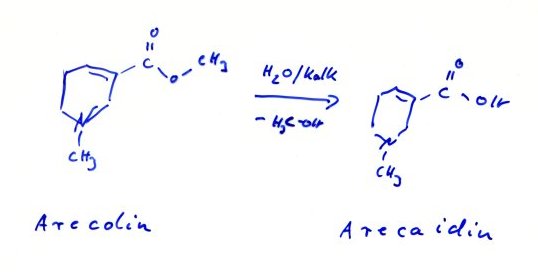
muss unsere Vorfahren für ihre Phantasie bewundern. Schon als Student hat mich das bekannte Beispiel der
Betel-Kauer[5]
beeindruckt : Wie konnte man nur, bereits um vor 500 v.Chr., auf die
Idee kommen Arecanüsse, in denen das Alkaloid Arecaidin als Arecolin verestert vorliegt, mit ungelöschtem Kalk zur Verseifung des
Esters zu kauen und dem Ganzen, zur Übertönung des wiederlichen Kalkgeschmack,
auch noch die Scharfstoffdroge "piper betle" zuzusetzen ? Und alles
rein empirisch gefunden, nur um sich ein Rauscherlebnis zu verschaffen? Aber sogar für die antibiotischen Eigenschaften von
Schimmelpilzen gebührt eigentlich nicht Alexander Flemming das Erstentdeckungsrecht.
Die mongolischen Reitervölker verwendeten schon vor Jahrhunderten auf unter
"exakten Kulturbedingungen", auf Pferdesattelleder in Gruben mit
Pferdemist wachsende Schimmelpilze für Wundverbände[6]. Auch bekannte Nahrungspflanzen wie Zwiebeln und Knoblauch
wurden nachweislich schon 3000 v.Chr. medizinisch verwendet. Aus dem Jahre 1600
v. Chr. ist dann ein Streik der Pyramidenarbeiter aktenkundig[7].
Man hatte ihnen keinen Knoblauch, der gegen die, aufgrund der einseitigen
Ernährung verbreiteten, Verdauungsbeschwerden angewendet wurde, zugeteilt. Der Wirkmechanismus dieser Gemüse ist heute bekannt.
Aus den in diesen Pflanzen vorkommenden Glucosinolaten werden durch
biochemische Prozesse die mit breitem antibiotischem Wirkungsspektrum
versehenen Isothiocyanate freigesetzt.
Doch wie entwickelt man chemische definierte
Arzneistoffe ?
E.Kutter schrieb 1978 in seinem Buch
"Arzneimittelentwicklung" eine Einschätzung, die im wesentlichen bis
heute richtig ist, auch wenn seither natürlich Fortschritte gemacht wurden :
Am Anfang steht seit dem Beginn der
Arzneimittelforschung die "SCHÖPFERISCHE HYPOTHESE".
Einer der für den synthetisch arbeitenden Arzneimittelchemiker
gangbarer Wege ist dabei der Ansatz, bekannte chemische Reaktivität für biologische
Wirkungen zu nutzen.
Farbdia
Dies ist die Darstellung des Computers in einer
umgewöhnlichen Perspektive, gleichsam "von unten".
Dia.
B6 [Hexamethylentetramin Wirkung]
Dies ist die erste "designed drug", das
heißt, aufgrund chemischer Vorstellungen entwickelte Arzneisubstanz, lange vor
der Entdeckung des heute modernen, oder sollte ich sagen modischen,
"molecular modeling". Die zugrundeliegende Überlegung war, die pH abhängige
Spaltung der Aminalstruktur zur Freisetzung des Formaldehyd im Urin zur
"Desinfektion" der Harnwegsorgane zu nutzen. Über Ansäuern des Urins,
z.B. durch Gabe von Citronensäure, sollte dabei die Wirkung steuerbar sein.
Dia.
B7 [Arsphenamin Wirkung]
durch Paul Ehrlich [+ Hata Nobelpreis f.Med. 1908].
Wie bekannt, gründete Ehrlich seine Überlegungen auf zwei Tatsachen. Schon als
Student hatte ihn beeindruckt, daß es möglich war, durch Farbstoffe bestimmte
Teile von Gewebeschnitten selektiv anzufärben. In seiner Doktorarbeit über
"Beiträge zur Theorie und Praxis der histologischen Färbung"
vertiefte er dieses Interessengebiet.
Bereits im dritten Semester seines Medizinstudiums
hatte er sich bei der Beschäftigung mit einer Arbeit von Heubel über
Bleivergiftungen den zweiten Ursprung seiner Theorie über die chemische
Affinität zwischen Gift und Gewebszellen angeeignet. Ehrlichs Arbeiten
beeinflussten die Vorstellungen über Chemotherapeutica bis heute nachhaltig.
Dia. B8 [Prontosil Wirkung]
durch Gerhard Domagk[10] [Nobelpreis
Med. 1939] und Mitarbeiter. Die Auswirkungen, auch in den anderen
Arzneistoffklassen, sind bis heute in
unserem Arzneischatz zu finden. Auch diese Chemotherapeutica basierten auf der von
Ehrlich begründeten Auswertung selektiver Anfärbemethoden. Gleichzeitig liegt
hier der interessante Fall eines "falsch-positiven" Ergebnisses vor.
Nicht die Azostruktur, sondern das in vivo entstehende 4-Aminobenzolsulfonamid
war wirkentscheidend, was Domagk und Mitarbeiter jedoch bald erkannten. Vom Ansatz her lag hier ein "idealer"
Arzneistoff vor. Das 4-Aminobenzolsulfonamid verdrängt bekanntlich die
4-Aminobenzoesäure in der vom Bakterium durchgeführten Synthesekaskade zur
Folsäure. Die resultierende Schädigung macht dann
die Bakterien für das menschliche Immunsystem überwindbar. Da die menschliche
Zelle Folsäure nicht selbst synthetisiert, sondern als "Vitamin" von
außen aufnimmt, ist dieses Wirkprinzip für Menschen untoxisch. Von sonstigen Problemen [Resistenz] bei der Anwendung
von Sulfonamiden soll in dieser Betrachtungsweise dabei abgesehen werden. Für den Wirkmechanismus dieser Chemotherapeutica wurde
dann um 1960 von Albert[11]
der Begriff "pro-drug" geprägt, der heute noch viel benutzt wird. Er definierte ihn so: "Eine 'pro-drug'
ist eine Substanz, die nach der Applikation erst in die aktuelle Substanz
überführt wird und sich dann mit dem Rezeptor verbindet". Wichtige Erfolge gegen Infektionskrankheiten errangen
dann das Penicillin [ab 1929] und seine Nachfolger. Doch diesen Teil der
Entwicklung möchte ich, wie eingangs erwähnt, aussparen.
Die nächste mir bemerkenswerte Entdeckung sind die
ersten Versuche der Chemotherapie von Krebs. Die Geschichte der alkierenden Pharmaka gegen Krebs
beginnt mit der im ersten Weltkrieg gemachten Beobachtung, dass das
Vergiftungsbild von Gasvergifteten mit Schwefellost
mit Leukopenie[12]
einhergeht. Über den Stickstofflost [1940] wurde dann das zur
Leitsubstanz seiner Klasse werdende Cyclophosphamidi entwickelt.
Dia.
B9
[Cyclophosphamid Biolog.
Abbau, Wirkung]
Die Verbindung ist bei in vitro Tests nahezu unwirksam
und wird erst im Organismus in die Wirkform umgewandelt.
N,N-
Bis-(2-chlorethyl)-phosphorsäurediamid (IV), die "Wirkform",
neben anderen Metaboliten wie
4-Oxocyclophosphamid (V) oder Carbophosphamid
(VI).
Auch auf dem immer wichtiger werdenden Feld der Mittel
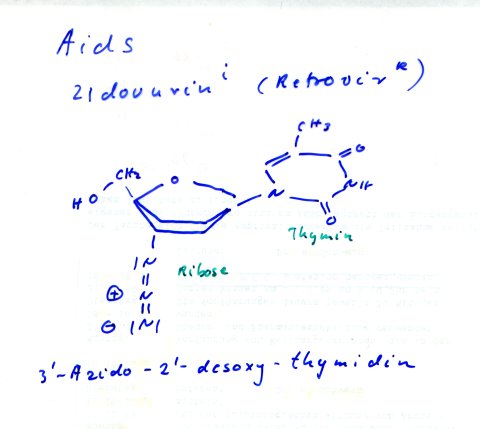
gegen Viren, Stichwort Aids[8],[13],
gibt es interessante neuere Ansätze. So gibt es, da Viren bekanntlich keinen
eigenen Stoffwechsel besitzen, nur wenige Angriffspunkt für eine Chemotherapie[9].
Man muss also den, wie ich glaube modernsten, aber auch schwierigsten Weg, über mögliche biochemischen Unterschiede zwischen "Eigen" und "Fremd" wählen, um zum Erfolg zu kommen. Ein Beispiel stellt die 1977 erstmals publizierte antiviral wirksame Substanz Acicloviri, dar.
Dia.
B10 [Aciclovir Wirkung] [Nucleosid-analogon mit
"falschem Zucker", d.h. einer nur rudimentären Seitenkette]
Wirkform ist das Aciclovirtriphosphat, welches durch
stufenweise enzymatische Phosphorylierung entsteht. Interessanterweise durch
die viruseigene Thymidin-Kinase und nicht, wie nach der Struktur
eigentlich zu erwarten, durch eine Purin-Nucleosid-Kinase. Acicloviri
ist dabei überwiegend ein Substrat für die virus-kodierte und nur
untergeordnet für die zelleigene Thyminkinase. Das hat zur Folge, daß nicht
infizierte Zellen kaum Aciclovirmonophosphat bilden. Dieses ist jedoch
Voraussetzung für die nachfolgende Triphosphatbildung durch zelleigene
Kinasen zur Wirkform Aciclovirtriphosphat und Aufnahme dieser falschen
Basen in die Nucleinsäure der Zellen. Dadurch werden nur die infizierten Zellen
getroffen, in virusfreien Zellen erfolgt fast keine Schädigung.
Dia. B10 [E605-E600 Matthies 146]
erläutern
der zu einer erheblichen Verstärkung der toxischen
Wirkung führt, erwünschte oder unerwünscht drastische Veränderungen der Wirkung
eintreten.
Dia. B11 [Benzodiazepine Roth 258]
erläutern
die zu gleichsinnig wirksamen oder gar
wirkungsstärkere Substanzen führen können.
Aus ihren Bearbeitung resultierten dann oft
eigenständige Arzneistoffeinführungen, die mitunter sogar therapeutisch
wertvoller als die Leitverbindung waren.
Dia.
B11 [Acetylaminofluoren "Giftungsreaktion"]
Aus dem 2-Acetylaminofluoren[14], einem wenig
toxischen Synthesezwischenprodukt, machen die Leberenzyme der meisten
Säugetiere, auch der Mensch, ein als Alkylants wirkendes carcinogenes Kation.
Pikanterweise fehlen dem Meerschweinchen, einem häufig genutzten Versuchstier,
die Enzyme für die N-Hydroxylierung. Daher zeigt 2-Acetylaminofluoren bei ihm keine
krebserzeugende Wirkung. Füttert man jedoch das N-Hydroxylderivat, treten die,
nach den Erfahrungen am anderen Säugetieren zu erwartenden, Tumorraten auf. Ein augenfälliger Beweis, wie sorgfältig carcinogenes
Potential vor der Anwendung am Menschen experimentell ermittelt werden muss.
Welchen windungsreichen Weg Arzneistoffentwicklungen manchmal gehen, möchte ich
dadurch erläutern, daß ich auf das schon erwähnte Urotropini
zurückkomme. Erst in jüngster Zeit zeigte sich welche Möglichkeiten
noch in der ursprünglichen chemischen Idee stecken. Dazu musste man nur die Ausgangsidee mit moderneren Konzepten
verbinden. Um 1970 hatte Kreutzkamp[15] versucht,
die Transportformentheorie zu systematisieren.
Dia.
B12 [Transp. 1.ter Art und Transp. 2.ter Art]
Er definierte einerseits Transportformen 1.ter Art,
die dem Schema im oberen Teil des Dia entsprechen, also über Zerfall in Wirk-
und Transportteil ihre Wirkung entfalten. Allerdings können, wie z.B. beim Urotropin zu
beobachten, durch unspezifische Freisetzungen [von Formaldehyd] unerwünschte
Nebenwirkungen auftreten. Dieser Gefahr kann durch den Einsatz einer, nach
Kreutzkamp als Transportform 2.ter Art [unterer Teil des Dias] zu
bezeichnenden Weiterentwicklung, die direkt aus dem intakten Molekül den
Wirkteil auf den Rezeptor überträgt, ohne daß dieser frei
auftritt, vermieden werden. Ob den Entwicklern des Taurolidini
(TaurolinR) diese Überlegungen bekannt waren oder nicht, weiß ich leider
nicht.
Dia.
B13 [Taurolidin Wirkung]
Der von ihnen entwickelte Arzneistoff stellt jedoch
ein Musterbeispiel für eine Transportform 2.ter Art dar.
Hiermit bin ich fast am Ende meiner Ausführungen.
Ich hoffe, ich konnte Ihnen, ich erinnere nur an den
Weg von 1894 Urotropin bis 1989 Taurolidin, ausgehend von der
historischen Entwicklung einmündend in moderne Arbeit zeigen, daß das Prinzip chemische
Reaktivität zur Erzielung biologischer Wirkungen zu nutzen
zwar alt bekannt, aber immer noch als aktuell ist. Die Aufgabe des
Arzneimittelchemikers ist es, auf der Basis guter Chemischer Kenntnisse mit
erweitertem Biologischen Wissen die Wirkungen von Chemisch definierten
Substanzen auf biologische Systeme zu erforschen.
| [1] | Permeabilitätsbeeinflußung, Änderung des Redox-Potential, pKa-Wertänderungen |
| [2] | 1) DD-Carboxypeptidase spaltet D-Ala--Ala, 2) Transpeptidase verknüpft D-Ala mit Glycin |
| [3] | international nonproprietary name |
| [4] | Reaktion von Zellen auf Metallspuren aus OP-Besteck |
| [5] | Schutz durch MESNA |
| [6] |
1) Gesteigerter DNA-Reparaturmechanismus, 2) Permeabilitätseinschränkungen, 3) Produktion nucleophiler Stoffe als "Abfangreagentien" |
| [7] | Entartung best. Retikulumszellen des lymph.
Systems |
| [8] |
Zidovudin i,
Retrovir R |
| [9] |
1) Zellwandpenetration, 2) Uncoating (Abstoßen der Eiweißhülle), 3) Replikation (Virenbau), 4)Maturation (Neue Eiweißhülle), 5) Release (Freisetzung) |
| [1] | Neutrale Subst. erhalten den Namen des Moleküls. Nur anionische o |
| [2] | Roth / Fenner, Arzneistoffe, Thieme Verlag, S.206 (1988) |
| [3] | Ann. 51, 1 (1845) |
| [4] | B. Rosenberg, Nature, 205, 698 (1965) |
| [5] | E. Schneider, PhuZ, 6,161 (1986) |
| [6] | Literatur ? |
| [7] | J. Lutomski, PhuZ 2, 45 (1980) |
| [8] | Nicolaur |
| [9] | A.T. Shohl u. C.L. Deming, J.Urol., 4, 419, (1920) |
| [10] | G. Domagk |
| [11] | A. Albert,
Selectiv Toxicity, S.30 (1960) |
| [12] | Leukozytenuntergang unter 4000 Zellen/mm3. Anzeichen für Zelluntergang wie er z.B. beim Krebs vorkommt. |
| [13] | RetrovirR (Zidovudini) 3'-Azido-2'-desoxythymidin, hemmt die reverse Transkriptase der Retroviren |
| [14] | Naehrstedt, PhuZ, 5, 151 (1977) |
| [15] | wie 2 |
| [16] | P.G. Waser / E. Sibler, Inovative Appr. in Drug Res., 155 (1986) |
![Goldverbindungen [Auranofin]](89-schtag-2.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}